- 1
-
SNPRelate(Zheng et al., 2012) for GDS file handling and genotype data management (Zheng et al., 2012) - 2
-
GENESISfor genome-wide association analysis and relatedness estimation (Manichaikul et al., 2010; Conomos et al., 2015) - 3
-
GWASToolsfor genotype data management and quality control - 4
-
qqmanfor creating Q-Q and Manhattan plots
Lecture 05: GWAS in R
PUBH 8878, Statistical Genetics
Overview
This session walks through a minimal GWAS workflow in R using SNPRelate and GENESIS. There exist many tools for GWAS, including PLINK, BOLT-LMM, REGENIE, SAIGE, and others. Here, we focus on GENESIS to illustrate the key steps.
We work with a small example GDS bundled with GENESIS to keep runtime short while illustrating the full pipeline:
- Quality control (variant- and sample‑level)
- LD pruning for structure/relatedness tasks
- Relatedness (KING‑robust) and population structure (PC‑AiR)
- Null model fitting (linear mixed model)
- Single‑variant association and diagnostics (QQ, λGC)
Setup
Step 1: Accessing and Loading Data
HapMap 3
- HapMap3 is an integrated reference of common and low-frequency alleles. The project genotyped 1.6 million common SNPs in 1,184 reference individuals from 11 global populations. (The International HapMap 3 Consortium, 2010)
- The project mapped linkage disequilibrium (LD) patterns used for tag‑SNP selection and served as an early imputation reference. (The International HapMap 3 Consortium, 2010; The International HapMap Consortium, 2005)
- Here, we will use a small subset from ASW (African ancestry in the Southwest USA) and MXL (Mexican ancestry in Los Angeles) samples
1gdsfile <- system.file("extdata", "HapMap_ASW_MXL_geno.gds", package = "GENESIS")
2g <- SNPRelate::snpgdsOpen(gdsfile)
3samp.id <- read.gdsn(index.gdsn(g, "sample.id"))
snp.id <- read.gdsn(index.gdsn(g, "snp.id"))
chr <- read.gdsn(index.gdsn(g, "snp.chromosome"))
pos <- read.gdsn(index.gdsn(g, "snp.position"))- 1
- Locate the example GDS file included with the GENESIS package
- 2
- Open the GDS file for random access to genotype data
- 3
- Read sample IDs, SNP IDs, chromosome numbers, and positions from the GDS file
We can also download the associated metadata
metadata <- readr::read_tsv("https://ftp.ncbi.nlm.nih.gov/hapmap/genotypes/hapmap3_r3/relationships_w_pops_041510.txt", show_col_types = FALSE)
head(metadata)# A tibble: 6 × 7
FID IID dad mom sex pheno population
<chr> <chr> <chr> <chr> <dbl> <dbl> <chr>
1 2427 NA19919 NA19908 NA19909 1 0 ASW
2 2431 NA19916 0 0 1 0 ASW
3 2424 NA19835 0 0 2 0 ASW
4 2469 NA20282 0 0 2 0 ASW
5 2368 NA19703 0 0 1 0 ASW
6 2425 NA19902 NA19900 NA19901 2 0 ASW metadata |>
mutate(population = case_when(population == "MEX" ~ "MXL",
TRUE ~ population))# A tibble: 1,397 × 7
FID IID dad mom sex pheno population
<chr> <chr> <chr> <chr> <dbl> <dbl> <chr>
1 2427 NA19919 NA19908 NA19909 1 0 ASW
2 2431 NA19916 0 0 1 0 ASW
3 2424 NA19835 0 0 2 0 ASW
4 2469 NA20282 0 0 2 0 ASW
5 2368 NA19703 0 0 1 0 ASW
6 2425 NA19902 NA19900 NA19901 2 0 ASW
7 2425 NA19901 0 0 2 0 ASW
8 2427 NA19908 0 0 1 0 ASW
9 2430 NA19914 0 0 2 0 ASW
10 2470 NA20287 0 0 2 0 ASW
# ℹ 1,387 more rowsStep 2: Pre-Modeling QC
Genotype quality control
Filtering for minor allele frequency (MAF)
- SNP-chips often determine genotypes based on intensities for each allele
- When MAF is low, genotype clusters can overlap, leading to genotyping errors
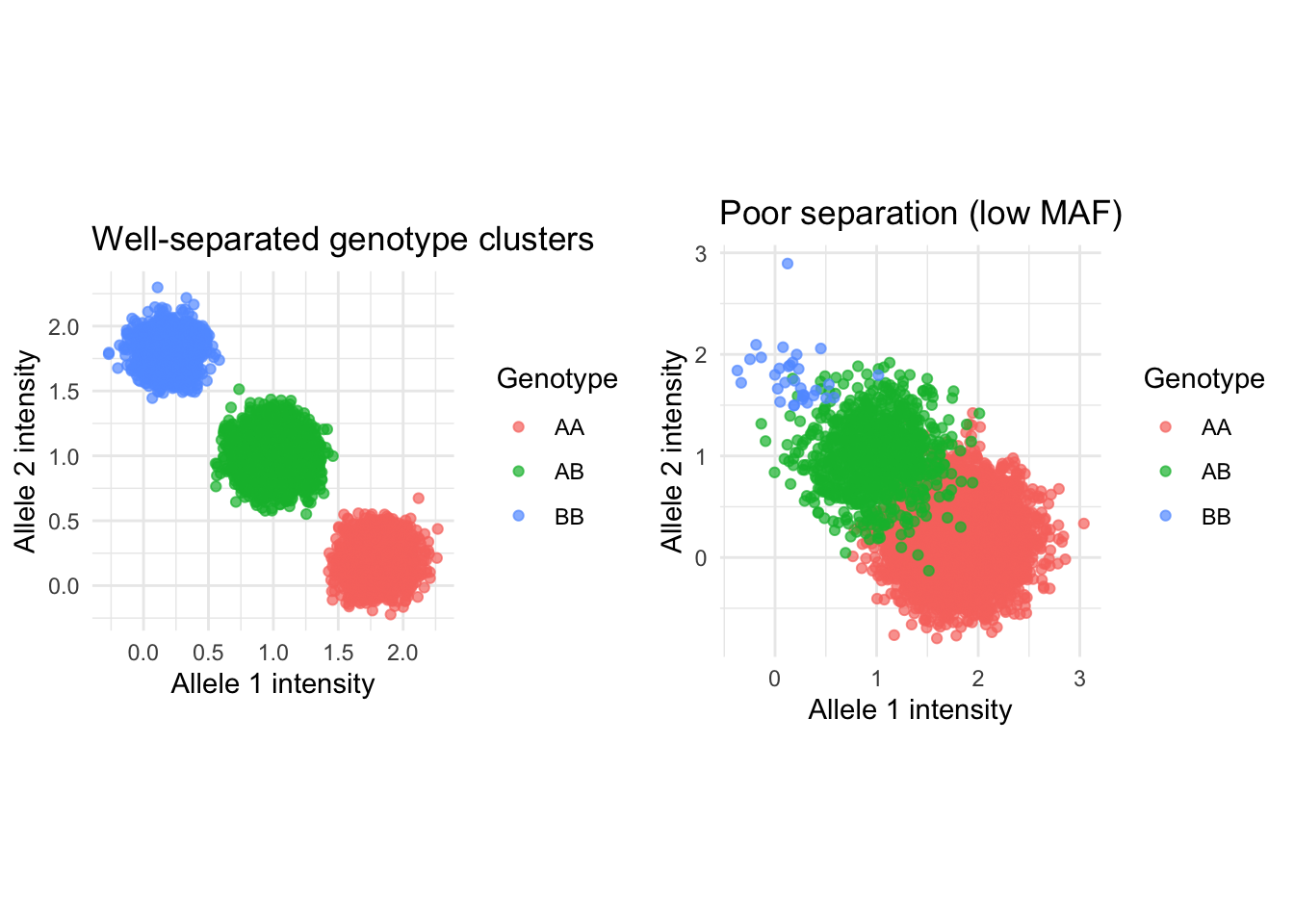
We can compute various statistics regarding allele frequency using snpgdsSNPRateFreq
af <- snpgdsSNPRateFreq(g)
str(af)
maf <- af$MinorFreqList of 3
$ AlleleFreq : num [1:20000] 0.39 0.494 0.101 0.486 0.445 ...
$ MinorFreq : num [1:20000] 0.39 0.494 0.101 0.486 0.445 ...
$ MissingRate: num [1:20000] 0 0 0 0 0.00578 ...We can see that af contains both minor allele frequency and missing rate per SNP.
data.frame(maf = maf) |>
mutate(maf_le05 = maf < 0.05) |>
ggplot(aes(x = maf, fill = maf_le05)) +
geom_histogram(binwidth = 0.01, color = "black") +
geom_vline(xintercept = c(0.05), linetype = "dashed", color = "red") +
theme_minimal() +
viridis::scale_fill_viridis(discrete = TRUE, end = .9, begin = .2) +
labs(fill = "MAF < .05")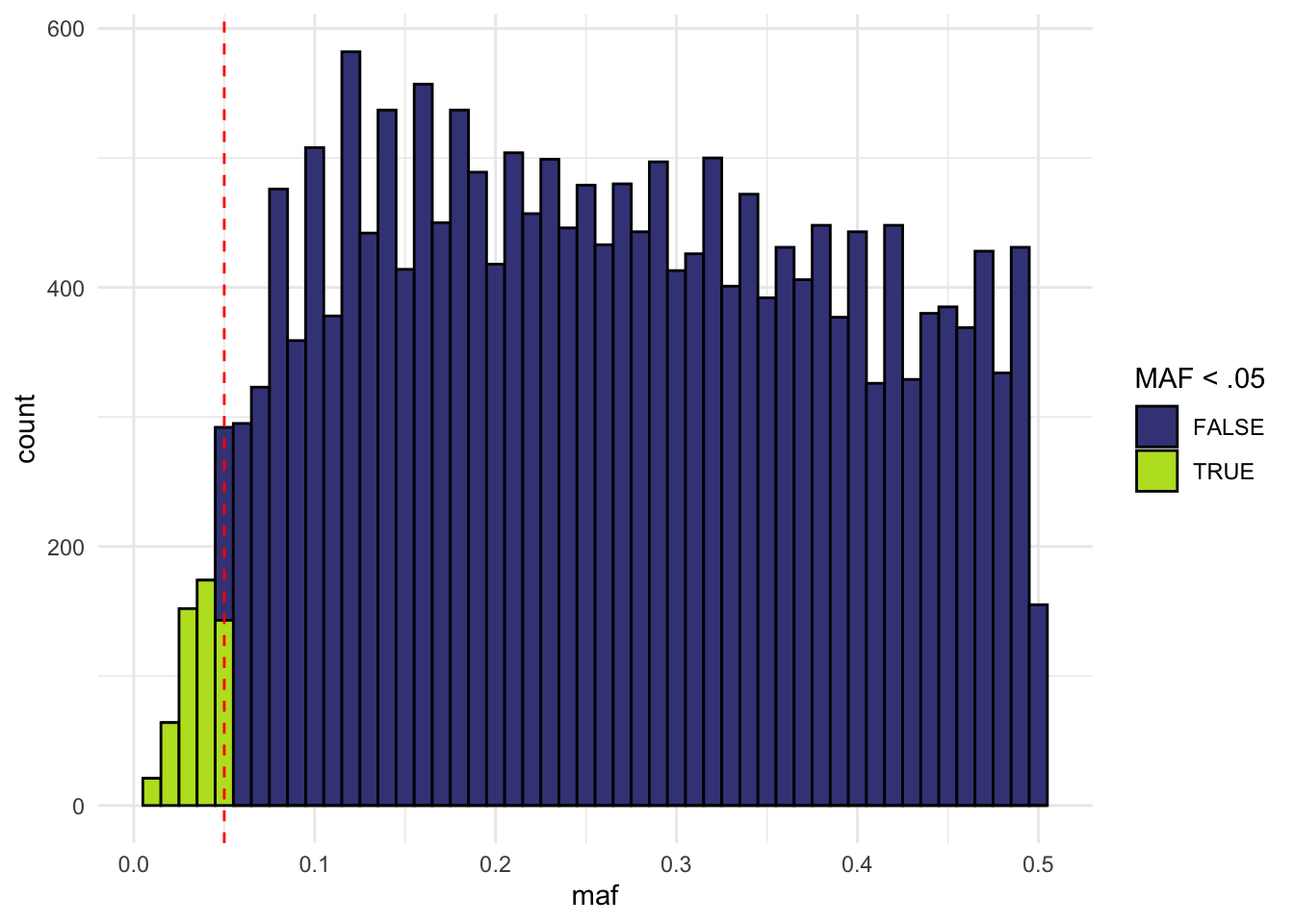
Variant missingness
- Missingness can indicate genotyping errors or batch effects
- Missingness can be non-random with respect to phenotype or ancestry
- Typically filter variants with > 2% missingness
- We can use the missingness results from our
afobject - Additionally, we can retrieve the genotype matrix itself, and manually compute the rate per SNP
G <- snpgdsGetGeno(g, with.id = TRUE)Genotype matrix: 20000 SNPs X 173 samplesgeno_mat <- G$genotype
dim(geno_mat)[1] 20000 173Indexing in R goes rows-by-columns. So, For geno_mat, the SNPs are rows and the samples are columns. We can inspect subset of the matrix:
geno_mat[10:20, 1:10] [,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] [,9] [,10]
[1,] 2 0 0 1 0 1 0 1 2 0
[2,] 0 1 1 1 1 0 0 0 2 0
[3,] 1 1 1 1 0 0 1 0 2 1
[4,] 0 0 0 0 0 0 0 0 0 1
[5,] 0 0 0 0 0 0 0 0 0 0
[6,] 1 0 1 1 0 0 0 2 1 0
[7,] 2 1 1 1 0 0 0 2 0 1
[8,] 0 0 0 0 1 1 0 0 2 0
[9,] 1 0 0 NA 0 1 0 2 0 0
[10,] 1 1 0 1 1 1 1 0 0 0
[11,] 1 0 1 0 1 1 0 2 1 0SNPRelate allows for four possible values stored in the genotype matrix: 0, 1, 2, 3. Consider a bi-allelic SNP site with possible alleles A and B.
- 0 indicates BB
- 1 indicates AB
- 2 indicates AA
- 3 indicates a missing genotype
For multi-allelic sites, it is a count of the reference allele.
miss_var_manual <- rowMeans(is.na(geno_mat))
miss_var <- af$MissingRate
identical(miss_var, miss_var_manual)[1] TRUEdata.frame(miss_var = miss_var) |>
mutate(miss_var_gt02 = miss_var > 0.02) |>
ggplot(aes(x = miss_var, fill = miss_var_gt02)) +
geom_histogram(binwidth = 0.001, color = "black") +
geom_vline(xintercept = c(0.02), linetype = "dashed", color = "red") +
theme_minimal() +
viridis::scale_fill_viridis(discrete = TRUE, end = .9, begin = .2) +
labs(fill = "Missing > .02")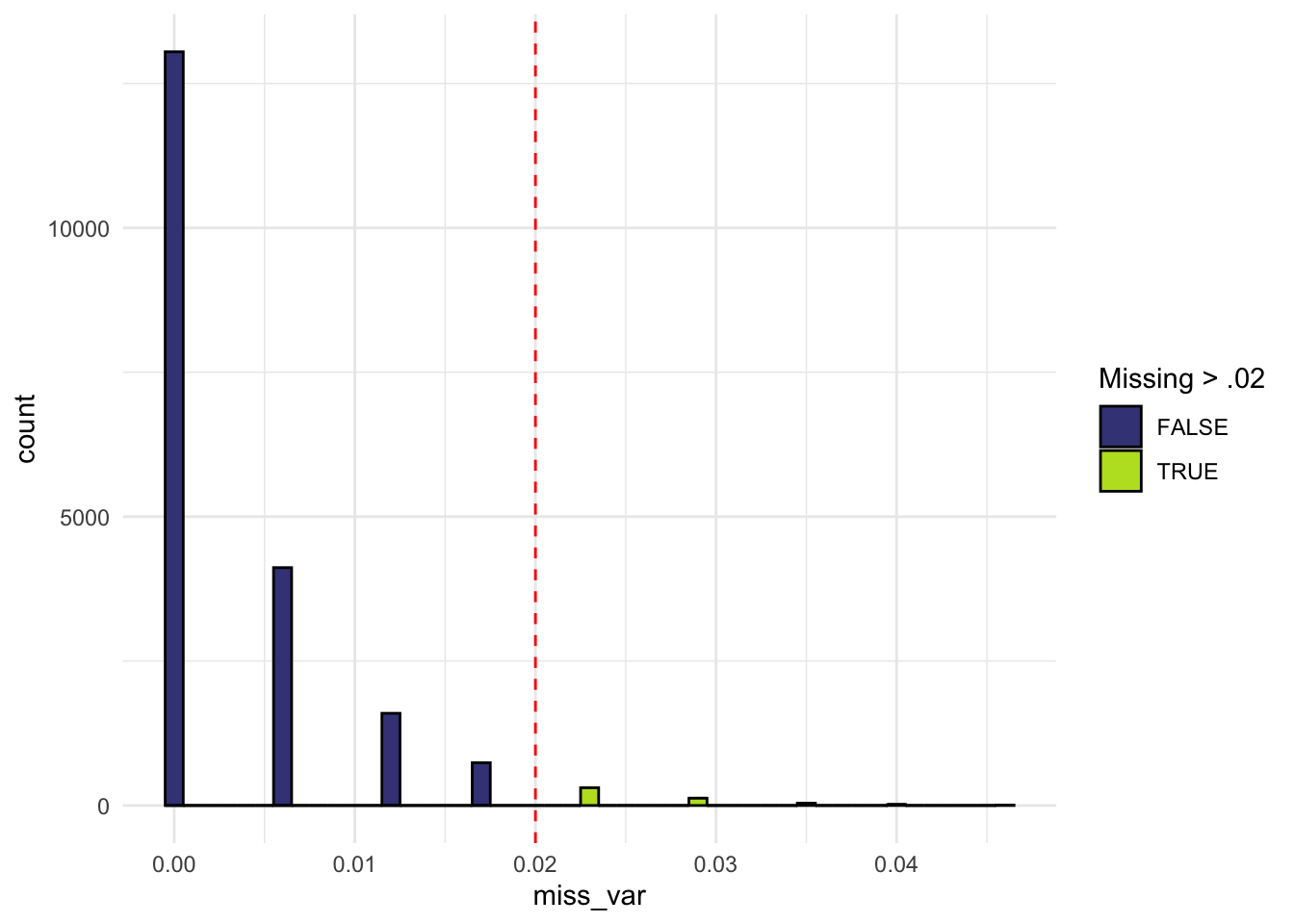
Sample-level missingness & heterozygosity
- Samples with high missingness may have poor DNA quality or technical issues
- Samples with extreme heterozygosity rates may be contaminated or mislabelled
samp_miss <- snpgdsSampMissRate(g)
data.frame(samp_miss = samp_miss) |>
mutate(samp_outlier = samp_miss > 0.02) |>
ggplot(aes(x = samp_miss, fill = samp_outlier)) +
geom_histogram(binwidth = 0.01, color = "black") +
geom_vline(xintercept = c(0.02), linetype = "dashed", color = "red") +
theme_minimal() +
labs(title = "Sample missingness", x = "Missing rate")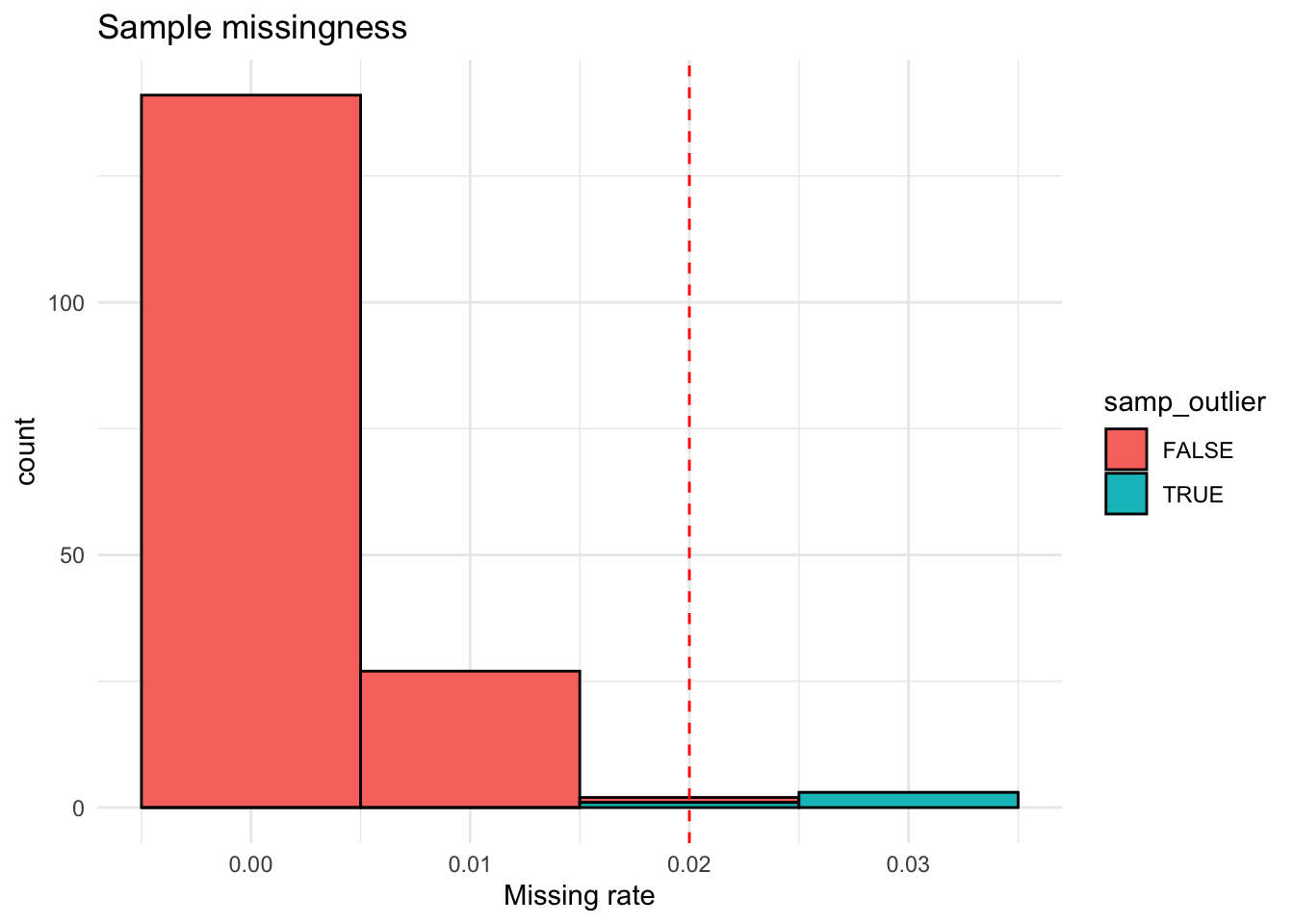
het_rate <- colMeans(geno_mat == 1, na.rm = TRUE)
data.frame(het_rate = het_rate) |>
mutate(het_outlier = abs(scale(het_rate)) > 3) |>
ggplot(aes(x = het_rate, fill = het_outlier)) +
geom_histogram(binwidth = 0.01, color = "black") +
geom_vline(xintercept = c(mean(het_rate) + 3 * sd(het_rate),
mean(het_rate) - 3 * sd(het_rate)),
linetype = "dashed", color = "red") +
theme_minimal() +
labs(title = "Heterozygosity rate", x = "Heterozygosity rate")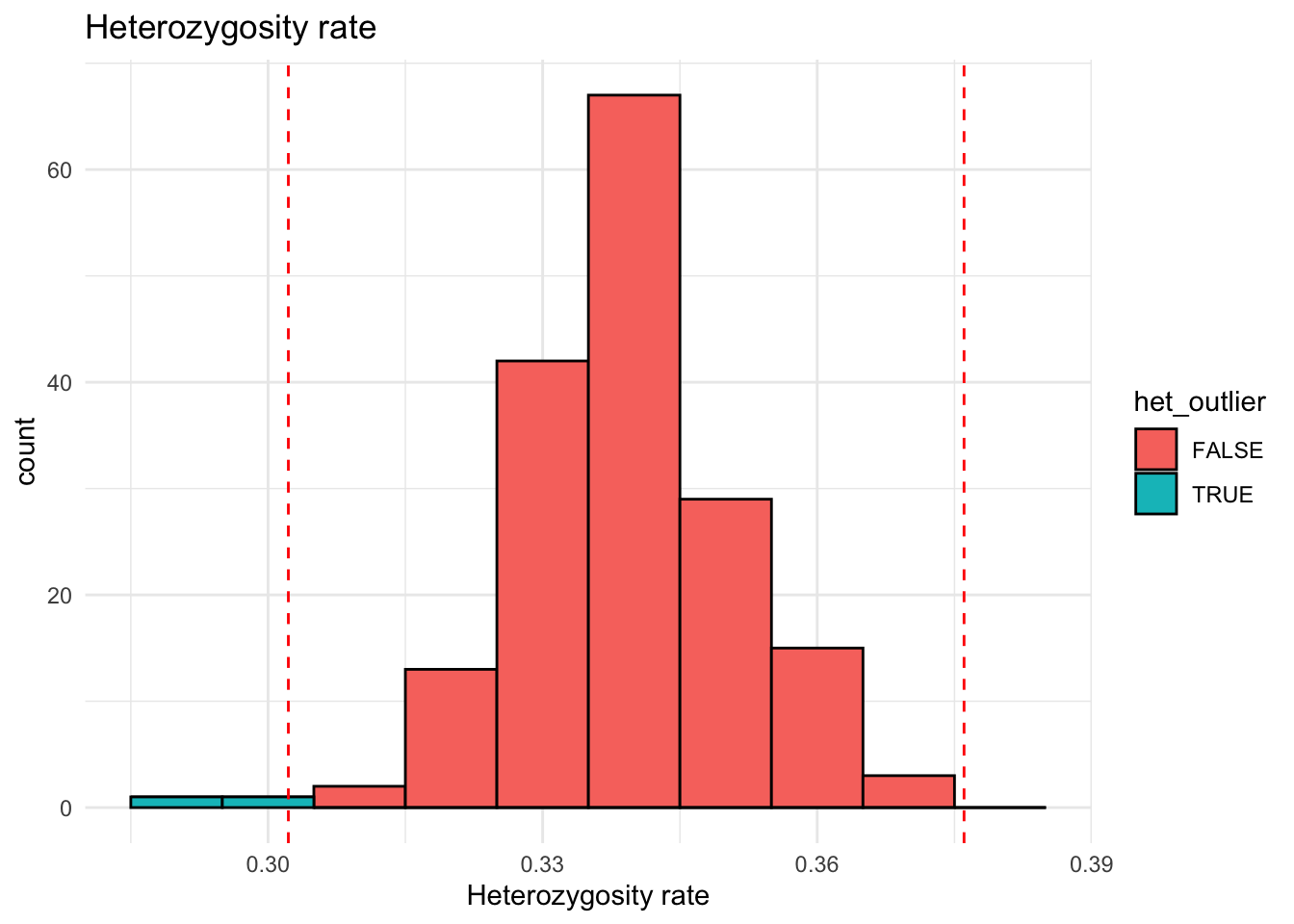
Thresholding
Putting it together: below we (i) filter samples on missingness/heterozygosity, (ii) apply preliminary variant filters (missingness + MAF), (iii) LD‑prune and estimate relatedness (KING) on that prelim set, and later (iv) compute HWE within ancestry‑homogeneous unrelated subsets using the metadata to finalize variant inclusion. This order avoids false HWE failures from population mixture/relatedness. (Anderson et al., 2010)
thr_samp_miss <- 0.02
thr_var_miss <- 0.02
thr_maf <- 0.05
thr_het_z <- 3
keep_sample <- which(samp_miss <= thr_samp_miss & abs(scale(het_rate)) < thr_het_z)
samp.keep.id <- samp.id[keep_sample]
keep_snp_prelim <- which(miss_var <= thr_var_miss & maf >= thr_maf)
snp.prelim.id <- snp.id[keep_snp_prelim]
length(snp.prelim.id)[1] 18960SNP correlation structure
max_plot_snps <- 100
ld_snp_ids <- snp.prelim.id[seq_len(max_plot_snps)]
ld_mat <- snpgdsLDMat(
g,
sample.id = samp.keep.id,
snp.id = ld_snp_ids,
slide = -1,
method = "corr"
)Linkage Disequilibrium (LD) estimation on genotypes:
# of samples: 167
# of SNPs: 100
using 1 thread
method: correlation
LD matrix: the sum of all selected genotypes (0,1,2) = 9659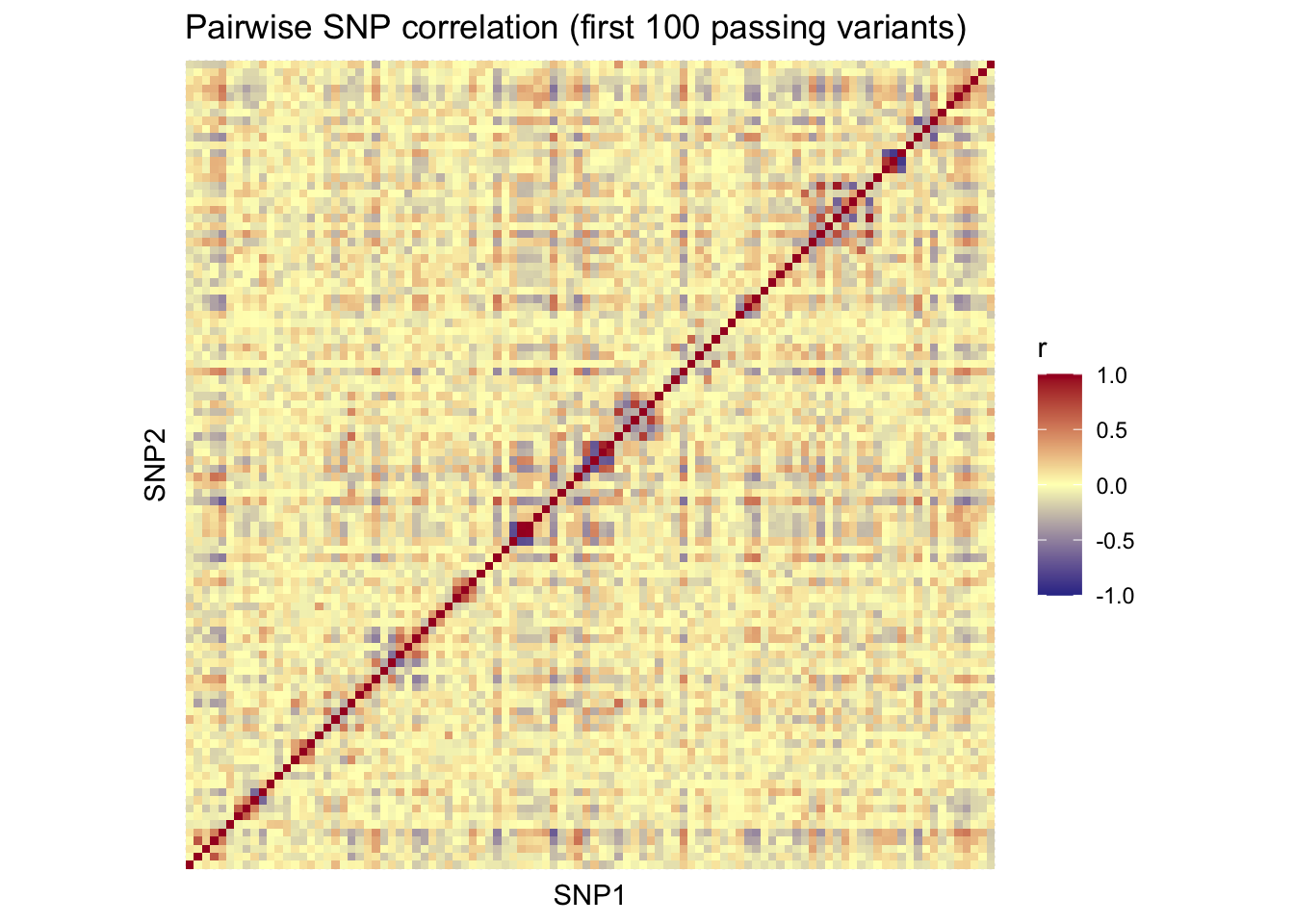
LD pruning
LD pruning removes variants in high LD to yield a subset of approximately independent variants. This is useful for population structure and relatedness estimation, which can be distorted by large blocks of correlated SNPs.
The algorithm works as follows (Zheng et al., 2012):
- Randomly select a starting position i (
start.pos="random"), i=1 ifstart.pos="first", or i=last ifstart.pos="last"; and let the current SNP set S={ i }; - For each right position j from i+1 to n: if any LD between j and k is greater than
ld.threshold, where k belongs to S, and both of j and k are in the sliding window, then skip j; otherwise, let S be S + { j }; - For each left position j from i-1 to 1: if any LD between j and k is greater than
ld.threshold, where k belongs to S, and both of j and k are in the sliding window, then skip j; otherwise, let S be S + { j }; - Output S, the final selection of SNPs.
set.seed(1)
ld_prune <- snpgdsLDpruning(g,
sample.id = samp.keep.id,
snp.id = snp.prelim.id,
autosome.only = TRUE,
method = "r",
slide.max.bp = 500000,
ld.threshold = sqrt(.1)
)SNP pruning based on LD:
Excluding 1,040 SNPs (non-autosomes or non-selection)
Excluding 0 SNP (monomorphic: TRUE, MAF: 0.005, missing rate: 0.05)
# of samples: 167
# of SNPs: 18,960
using 1 thread
sliding window: 500,000 basepairs, Inf SNPs
|LD| threshold: 0.316228
method: R
Chrom 1: |====================|====================|
21.27%, 4,255 / 20,000 (Wed Oct 8 09:36:05 2025)
4,255 markers are selected in total.snps_pruned <- unlist(ld_prune, use.names = FALSE)Ancestry PCs
With a kinship matrix in hand, we estimate ancestry PCs to use as covariates. PC‑AiR computes PCs on a subset of unrelated individuals and projects relateds onto that space, providing robust structure estimates in the presence of relatedness (Conomos et al., 2015).
- 1
- We need to close the SNPRelate connection before opening with GWASTools
- 2
- Open the GDS file with GWASTools
- 3
- Create a GenotypeData object for analysis
- 4
- Run PC-AiR using the KING kinship matrix to identify unrelateds and compute PCs
Using kinobj and divobj to partition samples into unrelated and related setsWorking with 167 samplesIdentifying relatives for each sample using kinship threshold 0.0220970869120796Identifying pairs of divergent samples using divergence threshold -0.0220970869120796Partitioning samples into unrelated and related sets...Unrelated Set: 92 Samples
Related Set: 75 SamplesPerforming PCA on the Unrelated Set...Predicting PC Values for the Related Set...Principal Component Analysis (PCA) on genotypes:
Excluding 15,745 SNPs (non-autosomes or non-selection)
Excluding 0 SNP (monomorphic: TRUE, MAF: NaN, missing rate: NaN)
# of samples: 92
# of SNPs: 4,255
using 1 thread
# of principal components: 32
PCA: the sum of all selected genotypes (0,1,2) = 201672
CPU capabilities:
Wed Oct 8 09:36:05 2025 (internal increment: 5340)
[..................................................] 0%, ETC: ---
[==================================================] 100%, completed, 0s
Wed Oct 8 09:36:05 2025 Begin (eigenvalues and eigenvectors)
Wed Oct 8 09:36:05 2025 Done.
SNP Loading:
# of samples: 92
# of SNPs: 4,255
using 1 thread
using the top 32 eigenvectors
SNP Loading: the sum of all selected genotypes (0,1,2) = 201672
Wed Oct 8 09:36:05 2025 (internal increment: 42740)
[..................................................] 0%, ETC: ---
[==================================================] 100%, completed, 0s
Wed Oct 8 09:36:05 2025 Done.
Sample Loading:
# of samples: 75
# of SNPs: 4,255
using 1 thread
using the top 32 eigenvectors
Sample Loading: the sum of all selected genotypes (0,1,2) = 163562
Wed Oct 8 09:36:05 2025 (internal increment: 52428)
[..................................................] 0%, ETC: ---
[==================================================] 100%, completed, 0s
Wed Oct 8 09:36:05 2025 Done.pcair_tb <- pcair_out$vectors |>
as_tibble() |>
bind_cols("IID" = pcair_out$sample.id) |>
left_join(metadata, by = "IID")
pcair_tb |>
ggplot(aes(V1, V2, col = population)) +
geom_point()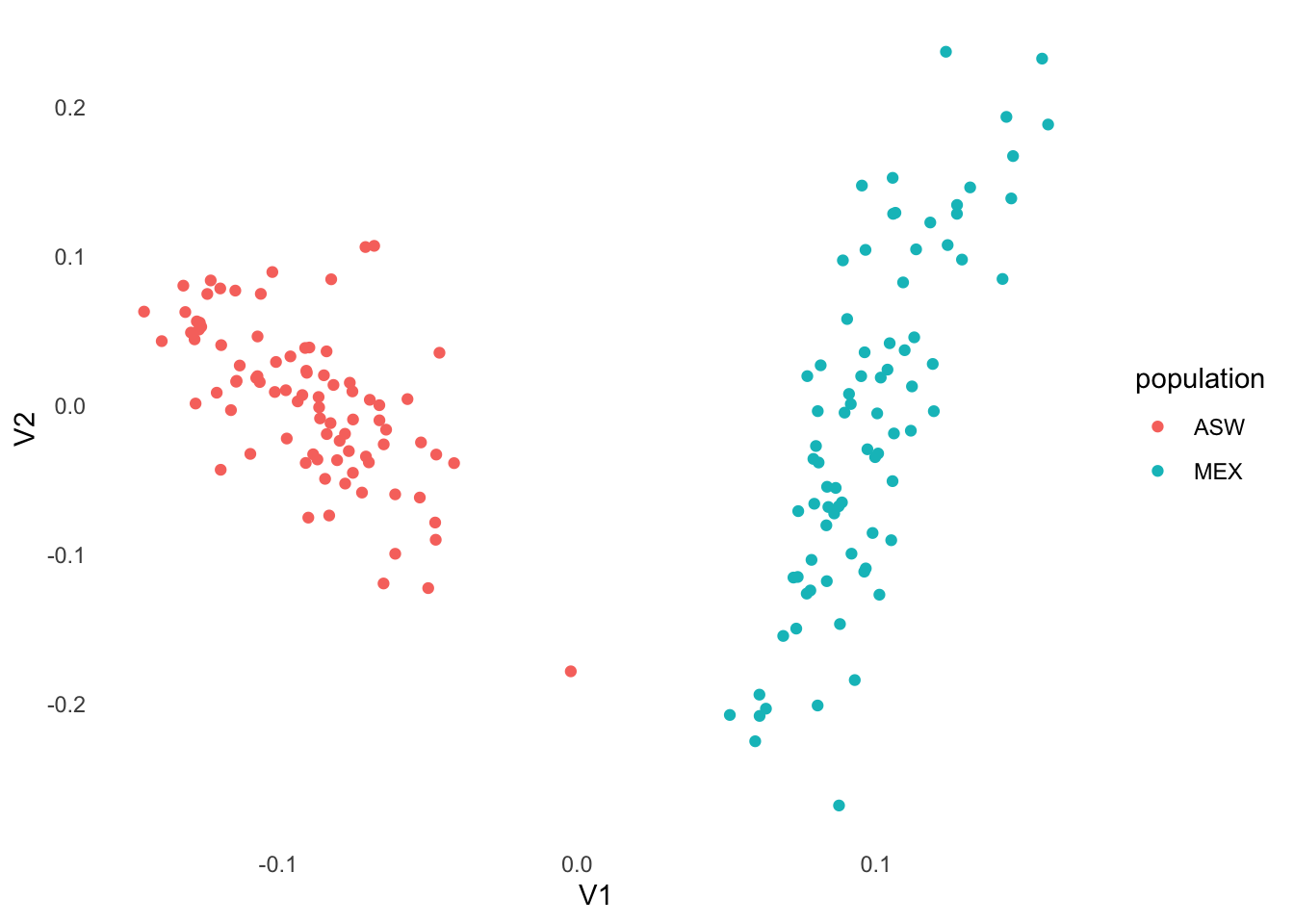
We can use a screeplot to visualize the variance explained by each PC. Depending on where we see an “elbow” in the screeplot, we might choose to include the corresponding PCs in our model.
pcair_out$values |>
as_tibble() |>
mutate(PC = row_number()) |>
ggplot(aes(PC, value)) +
geom_point() +
geom_line() +
scale_x_continuous(breaks = seq(1, 10, by = 1)) +
labs(title = "Screeplot of PC-AiR eigenvalues", x = "Principal Component", y = "Eigenvalue")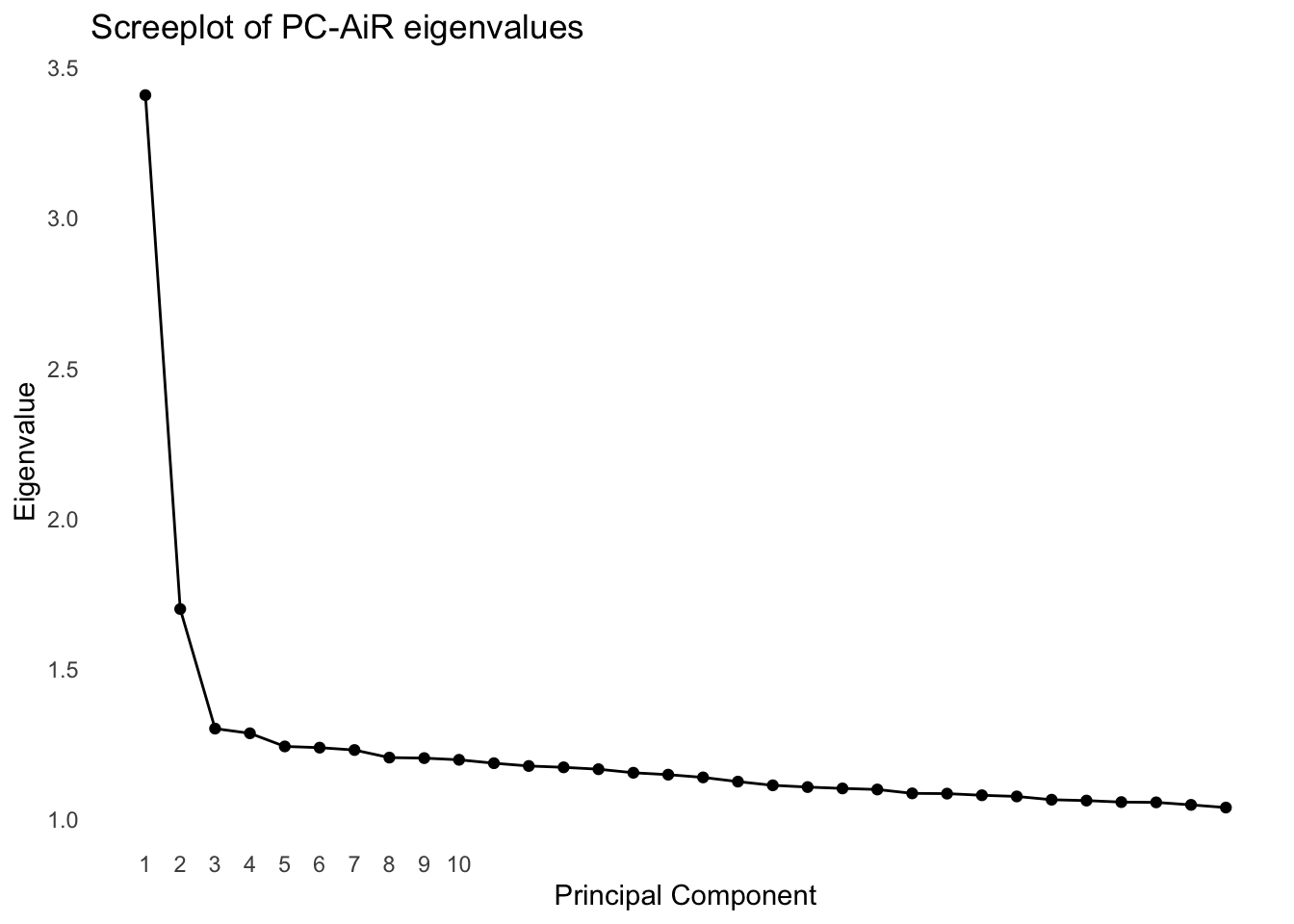
Construct phenotype and merge covariates
For the sake of this demo, we will simulate a quantitative trait with a known genetic effect
set.seed(99)
N <- length(samp.keep.id)
# Use recorded sex from metadata for covariates (single chromosome: no sex-chr analysis needed)
sex <- metadata |>
filter(IID %in% samp.keep.id) |>
mutate(sex = case_when(sex == 1 ~ "M",
TRUE ~ "F")) |>
pull(sex)
# Choose one moderately frequent SNP and simulate a quantitative trait with a planted effect
maf_keep <- maf[snp.keep.id]
ix <- which(!is.na(maf_keep) & maf_keep > 0.2 & maf_keep < 0.3)[1]
G1 <- GWASTools::getGenotypeSelection(geno_data, snpID = snp.keep.id[ix], scanID = samp.keep.id)
G1 <- as.numeric(G1)
y <- as.numeric(0.4* scale(G1) + 0.05 * (sex == "M") + rnorm(N))We now have a phenotype vector y and covariates. We need to construct a ScanAnnotationDataFrame object for the null model fitting.
pcs_df <- data.frame(
scanID = pcair_out$sample.id,
PC1 = pcair_out$vectors[, 1],
PC2 = pcair_out$vectors[, 2],
PC3 = pcair_out$vectors[, 3]
)
scanDF <- dplyr::left_join(
data.frame(scanID = samp.keep.id, y = y, sex = sex),
pcs_df,
by = "scanID"
)
scanAnnot2 <- GWASTools::ScanAnnotationDataFrame(scanDF)Step 3: Modeling
Null model
We first fit the mixed model under the null hypothesis that each SNP has no effect. The null includes covariates (age, sex, ancestry PCs) and a random genetic effect capturing relatedness; it does not include per‑SNP fixed effects.
The model:
y = X\beta + g + \epsilon
Where y is the phenotype, X\beta are fixed‑effect covariates, g \sim \mathcal{N}(0, \sigma^2_g K) is the random genetic effect with relatedness matrix K, and \epsilon \sim \mathcal{N}(0, \sigma^2_e I) is residual error.
Notes on estimation
- Covariance under the null: \operatorname{Var}(y) = V = \sigma^2_e I + \sigma^2_g K.
- We pass an ancestry‑adjusted GRM from PC‑Relate
- This yields diagonals near 1 (via
pcrelateToMatrix(scaleKin=2)) and controls for population structure in kinship estimation
- This yields diagonals near 1 (via
- Fixed effects (GLS) estimator given variance components: \hat{\beta} = (X^\top V^{-1} X)^{-1} X^\top V^{-1} y.
- Variance components for Gaussian outcomes are estimated by REML.
- Heritability estimate: h^2 = \sigma^2_g/(\sigma^2_g + \sigma^2_e).
nullmod <- GENESIS::fitNullModel(
scanAnnot2,
outcome = "y",
covars = c("sex", "PC1", "PC2", "PC3"),
cov.mat = pcrelMat, family = "gaussian"
)Computing Variance Component Estimates...Sigma^2_A log-lik RSS[1] 0.5717426 0.5717426 -243.7704375 1.0182557
[1] 0.1884426 0.8753057 -242.1753414 1.0461782
[1] 0.2420265 0.8685482 -242.1442635 1.0026855
[1] 0.2408589 0.8726307 -242.1442236 1.0000083
[1] 0.2411044 0.8724022 -242.1442228 1.0000000nullmod$varComp V_A V_resid.var
0.2411044 0.8724022 varComp reports the variance components from the mixed model. The entry V_A is the additive‑genetic variance captured by our GRM (PC‑Relate), and V_resid.var is the residual variance after adjusting for age, sex, and ancestry PCs. A convenient summary is the mixed‑model heritability on this working scale, h^2 = \frac{V_A}{V_A + V_{resid}}
varc <- nullmod$varComp
h2 <- unname(varc["V_A"] / sum(varc))
tibble::tibble(
component = c("V_A", "V_resid", "h2_mixed_model"),
value = c(varc["V_A"], varc["V_resid.var"], h2)
)# A tibble: 3 × 2
component value
<chr> <dbl>
1 V_A 0.241
2 V_resid 0.872
3 h2_mixed_model 0.217Association testing
We now test each SNP for association with the phenotype using a score test. The score test is computationally efficient because it only requires fitting the null model once, rather than refitting for each SNP as in a Wald or likelihood ratio test.
# Genotype iterator (block-wise scan of all SNPs kept by QC)
it <- GWASTools::GenotypeBlockIterator(
geno_data,
snpInclude = snp.keep.id,
snpBlock = 5000
)
assoc <- GENESIS::assocTestSingle(it, null.model = nullmod, test = "Score")Using 6 CPU coresres <- data.frame(
SNP = assoc$variant.id,
CHR = as.numeric(assoc$chr),
BP = assoc$pos,
P = assoc$Score.pval,
BETA = assoc$Est,
SE = assoc$Est.SE
)Step 4: Post‑GWAS
Extracting results
SNP BP BETA SE P
6 6 6 0.8396265 0.1568205 8.600221e-08
4628 4892 4892 -0.5765968 0.1501810 1.233630e-04
9363 9895 9895 -0.5772324 0.1532537 1.655421e-04
1796 1878 1878 -0.5223076 0.1415787 2.249887e-04
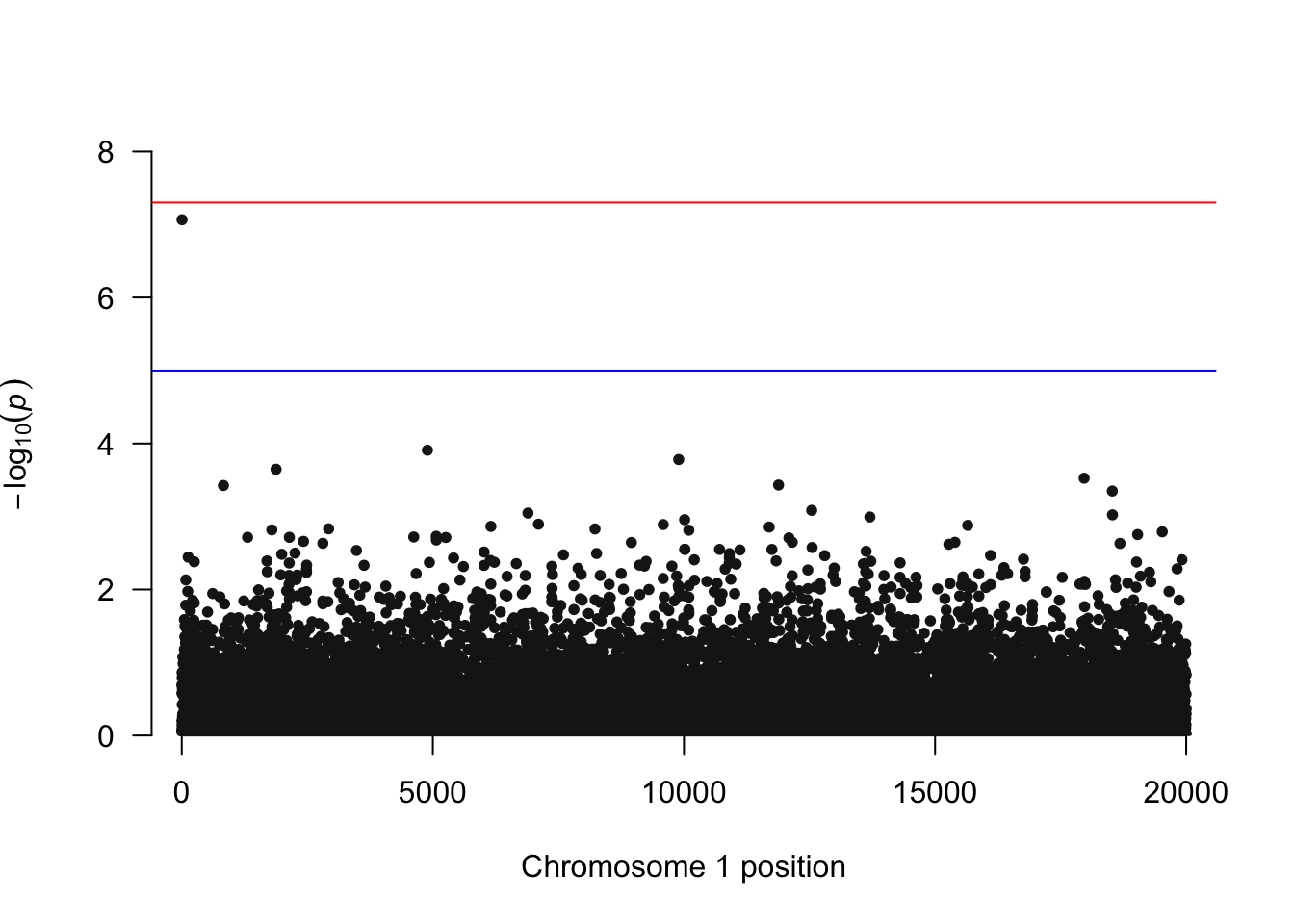
17050 17968 17968 0.4745057 0.1312115 2.987953e-04[1] 6Note that our top SNP is the one we simulated to have an effect.
Manhattan plot
We visualize the genome‑wide p-values using a Manhattan plot. The blue line indicates a p-value threshold of 1 \times 10^{-5}. Note that this is equivalent to a Bonferroni correction for 5,000 independent tests. The red line at 5 \times 10^{-8} is a common genome‑wide significance threshold for GWAS, though it is conservative for smaller studies or those with fewer variants.
# qqman expects columns named: CHR, BP, SNP, P
qqman::manhattan(
res,
chr = "CHR", bp = "BP", snp = "SNP", p = "P",
suggestiveline = -log10(1e-5)
)
QQ Plot
We compare observed p‑values to the null expectation to assess calibration. Few true signals should deviate strongly.
qqman::qq(res$P)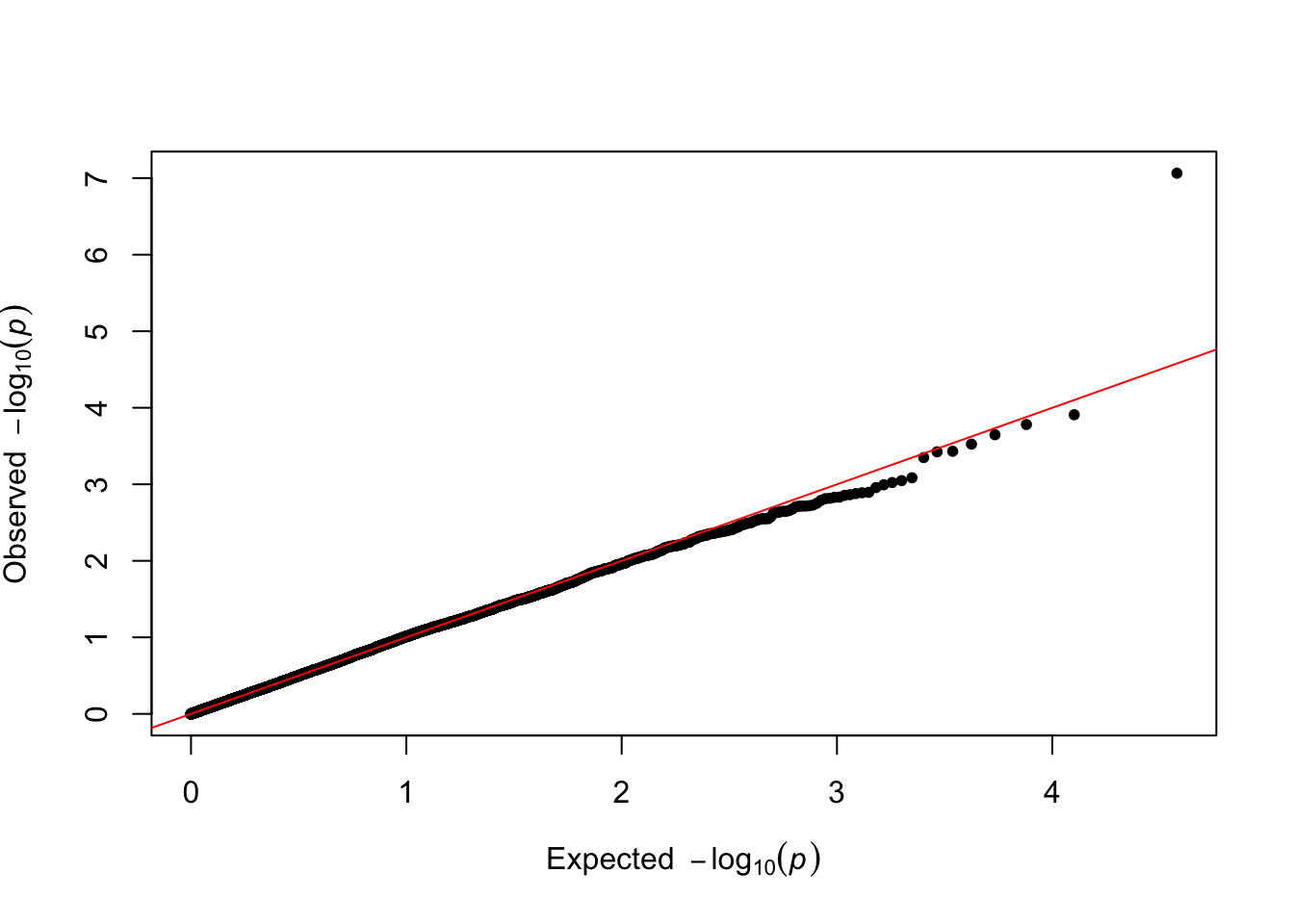
Genomic-control inflation factor
The genomic-control inflation factor is a single-number summary (\lambda_{GC}) of how inflated or conservative the genome‑wide test statistics are relative to the null. Values near 1 indicate well‑calibrated tests, >1 suggests inflation (e.g., population structure, relatedness, batch), <1 suggests conservative/underpowered tests
For 1‑df tests, the null statistic follows \chi^2_1, whose median is qchisq(0.5, 1) \approx 0.456. We will convert our p‑values to \chi^2 statistics T_i = \texttt{qchisq}(1 − p_i, df = 1). Then \lambda_{GC} = \text{median}(T_i) / m_0.
lambda_gc <- median(stats::qchisq(1 - res$P, df = 1), na.rm = TRUE) / 0.456
lambda_gc[1] 0.9851403Cleanup
When done, close the GDS connection
GWASTools::close(geno_reader)When to use a different tool?
- For very large datasets (e.g., UK Biobank), specialized tools like
BOLT-LMM,REGENIE, orSAIGEmay be more efficient - For binary traits, consider tools that handle case-control imbalance and relatedness effectively, such as
SAIGE - For rare variant analysis, consider burden tests or SKAT implemented in packages like
SKATorseqMeta
Further reading
Session Info
sessionInfo()R version 4.5.1 (2025-06-13)
Platform: aarch64-apple-darwin20
Running under: macOS Tahoe 26.0.1
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.5-arm64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.5-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.1
locale:
[1] C.UTF-8/C.UTF-8/C.UTF-8/C/C.UTF-8/C.UTF-8
time zone: America/New_York
tzcode source: internal
attached base packages:
[1] stats graphics grDevices datasets utils methods base
other attached packages:
[1] readr_2.1.5 dplyr_1.1.4 ggplot2_3.5.2
[4] qqman_0.1.9 GWASTools_1.54.0 Biobase_2.68.0
[7] BiocGenerics_0.54.0 generics_0.1.4 GENESIS_2.38.0
[10] SNPRelate_1.42.0 gdsfmt_1.44.1
loaded via a namespace (and not attached):
[1] Rdpack_2.6.4 DBI_1.2.3 gridExtra_2.3
[4] sandwich_3.1-1 rlang_1.1.6 magrittr_2.0.3
[7] SeqVarTools_1.46.0 compiler_4.5.1 RSQLite_2.4.3
[10] mgcv_1.9-3 reshape2_1.4.4 vctrs_0.6.5
[13] stringr_1.5.1 quantreg_6.1 pkgconfig_2.0.3
[16] shape_1.4.6.1 crayon_1.5.3 fastmap_1.2.0
[19] backports_1.5.0 XVector_0.48.0 labeling_0.4.3
[22] utf8_1.2.6 rmarkdown_2.29 tzdb_0.5.0
[25] UCSC.utils_1.4.0 nloptr_2.2.1 MatrixModels_0.5-4
[28] purrr_1.0.4 bit_4.6.0 xfun_0.52
[31] glmnet_4.1-10 jomo_2.7-6 logistf_1.26.1
[34] cachem_1.1.0 GenomeInfoDb_1.44.3 jsonlite_2.0.0
[37] blob_1.2.4 pan_1.9 BiocParallel_1.42.2
[40] broom_1.0.8 parallel_4.5.1 R6_2.6.1
[43] stringi_1.8.7 RColorBrewer_1.1-3 boot_1.3-31
[46] DNAcopy_1.82.0 rpart_4.1.24 lmtest_0.9-40
[49] GenomicRanges_1.60.0 Rcpp_1.1.0 iterators_1.0.14
[52] knitr_1.50 zoo_1.8-14 IRanges_2.42.0
[55] igraph_2.1.4 Matrix_1.7-3 splines_4.5.1
[58] nnet_7.3-20 tidyselect_1.2.1 viridis_0.6.5
[61] yaml_2.3.10 codetools_0.2-20 curl_6.2.3
[64] plyr_1.8.9 lattice_0.22-7 tibble_3.3.0
[67] quantsmooth_1.74.0 withr_3.0.2 evaluate_1.0.3
[70] survival_3.8-3 Biostrings_2.76.0 pillar_1.11.0
[73] BiocManager_1.30.26 mice_3.18.0 renv_1.1.5
[76] foreach_1.5.2 stats4_4.5.1 reformulas_0.4.1
[79] vroom_1.6.5 S4Vectors_0.46.0 hms_1.1.3
[82] scales_1.4.0 minqa_1.2.8 calibrate_1.7.7
[85] GWASExactHW_1.2 glue_1.8.0 tools_4.5.1
[88] data.table_1.17.4 lme4_1.1-37 SparseM_1.84-2
[91] cowplot_1.1.3 grid_4.5.1 tidyr_1.3.1
[94] rbibutils_2.3 nlme_3.1-168 formula.tools_1.7.1
[97] GenomeInfoDbData_1.2.14 cli_3.6.5 SeqArray_1.48.3
[100] viridisLite_0.4.2 gtable_0.3.6 digest_0.6.37
[103] operator.tools_1.6.3 farver_2.1.2 memoise_2.0.1
[106] htmltools_0.5.8.1 lifecycle_1.0.4 httr_1.4.7
[109] mitml_0.4-5 bit64_4.6.0-1 MASS_7.3-65 References
Anderson,C.A. et al. (2010) Data quality control in genetic case–control association studies. Nature Protocols, 5, 1564–1573.
Conomos,M.P. et al. (2016) Model-free estimation of recent genetic relatedness. The American Journal of Human Genetics, 98, 127–148.
Conomos,M.P. et al. (2015) Robust inference of population structure for ancestry prediction and correction of stratification in the presence of relatedness. Genetic Epidemiology, 39, 276–293.
Guo,S.W. and Thompson,E.A. (1992) Performing the exact test of Hardy–Weinberg proportion for multiple alleles. Biometrics, 48, 361–372.
Manichaikul,A. et al. (2010) Robust relationship inference in genome-wide association studies. Bioinformatics, 26, 2867–2873.
The International HapMap 3 Consortium (2010) Integrating common and rare genetic variation in diverse human populations. Nature, 467, 52–58.
The International HapMap Consortium (2005) A haplotype map of the human genome. Nature, 437, 1299–1320.
Wigginton,J.E. et al. (2005) A note on exact tests of Hardy–Weinberg equilibrium. The American Journal of Human Genetics, 76, 887–893.
Zheng,X. et al. (2012) A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics, 28, 3326–3328.